Treating with antioxidants early in Parkinson's disease process may halt degeneration and improve neuronal function
 of an intraneural Lewy-body in the Substantia nigra in Parkinson's disease. Credit: Wikipedia")
Northwestern Medicine scientists have identified a toxic cascade that leads to neuronal degeneration in patients with Parkinson's disease (PD) and figured out how to interrupt it, reports a study to be published September 7 in the journal Science.
Intervening with an antioxidant early in the disease process may break the degenerative cycle and improve neuron function in PD, the study showed.
The scientists also discovered that mouse models of PD didn't have the same abnormalities they found in human PD neurons, revealing the importance of studying human neurons to develop new therapies.
Dr. Dimitri Krainc, the Aaron Montgomery Ward Professor and chair of neurology at Northwestern University Feinberg School of Medicine, is the study senior author. Lena Burbulla, a postdoctoral fellow in Krainc's laboratory, is first author.
The research was started about six years ago in Krainc's lab at Massachusetts General Hospital and Harvard Medical School and was completed in the last four years at Feinberg.
PD is the second most common neurodegenerative disorder, primarily caused by the death of dopamine-containing neurons in the substantia nigra, a region of the brain involved in motor control. While people naturally lose dopamine neurons as they age, patients with PD lose a much larger number of these neurons and the remaining cells are no longer able to compensate.
Understanding how and why these neurons die is an important step in identifying treatments, Krainc said. While previous research indicated that the cellular mechanism behind the cell death involved the mitochondria and lysosomes, how these two pathways converge in dopamine neurons to cause cell death remained unknown up until now.
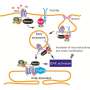
Using human neurons from Parkinson's patients, Krainc and colleagues identified a toxic cascade of mitochondrial and lysosomal dysfunction initiated by an accumulation of oxidized dopamine and a protein called alpha-synuclein. Specifically, the current study demonstrated that an accumulation of oxidized dopamine depressed the activity of lysosomal glucocerebrosidase (GCase), an enzyme implicated in PD. That depression in turn weakened overall lysosomal function and contributed to degeneration of neurons.
The accretion of oxidized dopamine didn't just interfere with lysosomes, however. Krainc and his colleagues discovered that the dopamine also damaged the neurons' mitochondria by increasing mitochondrial oxidant stress. These dysfunctional mitochondria led to increased oxidized dopamine levels, creating a vicious cycle.
"The mitochondrial and lysosomal pathways are two critical pathways in disease development," said Krainc, who also is the director of the Center for Rare Neurological Diseases and a professor of neurological surgery and of physiology. "Combined with the alpha-synuclein accumulation, this study links the major pathological features of PD."
Once they had catalogued this toxic cascade, Krainc and his colleagues began looking for ways to interrupt it.
"One of the key strategies that worked in our experiments is to treat dopamine neurons early in the toxic cascade with specific antioxidants that improve mitochondrial oxidant stress and lower oxidized dopamine," Krainc said. "With this approach, we found that we can attenuate or prevent the downstream toxic effects in human dopaminergic neurons."
This approach to interrupting the toxic cascade of oxidized dopamine may provide a target for the development of future therapies. However, identifying patients or subjects with early-stage neurodegeneration can be difficult, because damage has often occurred far before any symptoms are apparent, according to Krainc.
Consequently, genetic testing will be central to future diagnostic efforts. Causative genes are prime candidates for screening, while risk genes such as GBA1 are less conclusive but still important markers, Krainc said. Early detection will also rely on brain imaging and other clinical signifiers.
Interestingly, when compared to human cellular models, mouse models of PD did not demonstrate the same toxic cascade, according to the study. Krainc and his colleagues showed this is due to differences in metabolism of dopamine between species, and underscored the importance of studying human neurons to discover new targets for drug development.
More information: L.F. Burbulla el al., "Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease," Science (2017). science.sciencemag.org/lookup/ … 1126/science.aam9080