Global team enables child with a fatal genetic disease to recover

A young boy with a rare genetic disease that typically kills within weeks of birth is now 3 years old and in remission thanks to a collaborative effort that included physicians at King Saud University Department of Pediatrics in Riyadh, Saudi Arabia, and immunologists at the Icahn School of Medicine at Mount Sinai in New York.
A report published in the New England Journal of Medicine today describes how the global team combined exceptional supportive clinical therapy, genetic diagnosis and a novel immunotherapeutic drug known as a protein kinase inhibitor to bring the Saudi Arabian boy into full remission from his deadly disease, known as "USP18 deficiency" because it is caused by a mutation of the USP18 gene.
"The teamwork between our two institutions and others around the world is a textbook case of science without borders," says Dusan Bogunovic, Ph.D., Associate Professor of Microbiology, and Pediatrics, at the Icahn School of Medicine at Mount Sinai and co-corresponding author of the study. "We showed that even with a disease like USP18 deficiency, sound clinical care and timely drug administration can rescue patients from what was previously considered a death sentence."
USP18 (ubiquitin-specific peptidase 18) is a protein coding gene involved in immune system. It is important to regulate inflammation driven by a substance that our body normally secretes to fight off viruses, type 1 interferons. Mutations of USP18 result in an uncontrolled response to type 1 interferons, triggering IFN-I-mediated inflammation that's lethal in utero or shortly after birth. JAK1 inhibitor drugs, like ruxolitinib, the protein kinase inhibitor given to the Saudi Arabian boy, take over the intended role of USP18, and thus have the potential for a prompt and sustained recovery by patients.
Dr. Bogunovic and his lab, widely known for their work in the field of rare inflammatory diseases in children, first described USP18 deficiency in 2016. The following year, physicians at King Saud University reached out to Mount Sinai via Paris Descartes University in France about a gravely ill young patient in their intensive care unit who appeared to have a variant of the USP18 gene. Thus began a clinical/research collaboration—which included Paris Descartes University as well as Rockefeller University in New York—in which scientists characterized in detail the molecular basis of the disease through a battery of whole exome sequencing, expression assays, protein analysis, and antibody detection. "After seeing a potential variation in the USP18 gene, we conducted a complete set of tests to determine what it meant in terms of protein function," explains Marta Martin-Fernandez, Ph.D., a postdoctoral fellow at the Icahn School of Medicine and a first author of the published study, who performed many of its biochemical and genetic analyses. "Those findings confirmed for us that ruxolitinib was the appropriate treatment."
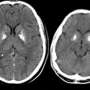
The young patient, who had been kept alive for months through the extraordinary care of physicians led by Fahad Alsohime, MD, Assistant Professor at the College of Medicine, King Saud University, was promptly put on oral, twice-daily doses of ruxolitinib. The dosage was increased after insufficient changes were seen, and within two weeks his symptoms began to rapidly improve, allowing doctors to wean him from respiratory support. Subsequent CT and MRI imaging showed a resolution of hemorrhaging, ischemia, cellulitis of the right forearm, and hydrocephalus, a condition in which cerebrospinal fluid accumulates in the brain. After two years of follow-up in an outpatient clinic in Riyadh, and continued administration of the JAK1 inhibitor, the child remains free of clinical problems and has been given an encouraging prognosis by his physicians. He will likely have to take ruxolitinib for the rest of his life.
The success of this case has provided a further springboard for Mount Sinai scientists to investigate the genomics and molecular/cellular biology behind conditions less severe than the boy's. This ongoing work links to other studies that have shown that JAK inhibitors—which were initially developed as anti-cancer drugs but proved to be largely ineffective—can improve symptoms and control disease activity in patients with other type 1 interferon abnormalities.
"We were able to demonstrate the benefits of rapid genetic diagnosis of an inherited disorder for which an immunosuppressant drug like ruxolitinib can provide effective and sustained treatment," says Dr. Bogunovic of Mount Sinai, a senior author on the publication. "That kind of discovery and drug repurposing must continue to be pursued by the scientific community without interruption."