New treatment for muscular dystrophy wins US regulatory approval

Research led by Professor Steve Wilton and Professor Sue Fletcher and licensed to Sarepta Therapeutics has delivered a second treatment for Duchenne muscular dystrophy, with the drug gaining accelerated approval by the U.S. Food and Drug Administration.
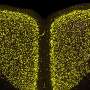
Mainly affecting boys, Duchenne muscular dystrophy (DMD) is an inherited disorder that causes progressive muscle weakness and loss of muscle mass. People with this disease are typically wheelchair dependent by age 12 and many do not survive beyond their mid-20s.
Although a rare disease, DMD is the most common of the childhood muscular dystrophies and was long considered untreatable. Collaborative research with Sarepta Therapeutics spanning two decades delivered the first approved molecular therapy for DMD in 2016.
Now, a second drug, golodirsen (Vyondys 53), has received accelerated approval by the U.S. Food and Drug Administration and while this means ongoing evaluation of treatment related benefits, the treatment becomes accessible to patients in the U.S..
Golodirsen was developed in the Perron Institute laboratory and licensed to Sarepta Therapeutics by The University of Western Australia. Prof. Wilton, Prof. Fletcher and their research team are now based at the Centre for Molecular Medicine and Innovative Therapeutics, a joint venture between Murdoch University and Western Australia's Perron Institute.
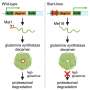
Both the DMD treatments developed by the Perth team exploit the cell machinery to trick cells into 'skipping' over the disease-causing error, acting as genetic 'whiteout." Boys receiving the exon skipping treatment in the United States have maintained the ability to walk into their mid- to late-teens, whereas without treatment, they would expect to be wheelchair bound around 12 years of age.
"The concept of tricking cells to skip genetic errors to treat Duchenne muscular dystrophy evolved during my development of diagnostic screening for neuromuscular diseases," Prof. Wilton said.
"Our group has been at the forefront in developing exon skipping as a therapy for Duchenne muscular dystrophy and this second approval is another huge step forward for the team and the treatment of this rare and fatal disease."
The breakthrough treatments are part of a larger body of work that describes compounds that could treat a large proportion of DMD sufferers.
"It's been estimated about 85 percent of Duchenne individuals could respond to this type of treatment, if all the potential drugs were developed," Prof. Wilton said.
One more drug, casimersen, is undergoing clinical evaluation for the treatment of another subset of DMD patients, increasing the number of patients who could benefit from exon skipping therapy. Each compound addresses different mutations in the dystrophin gene and the goal now is to see as many patients treated as possible.